
ISO15189中与试剂或消耗品有关的条文
检验试剂管理系统软件,符合ISO15189中有关试剂及消耗品的管理要求,相当多的包括最早一批通过15189认证的检验科都采用了检验试剂管理系统。
ISO15189中与试剂或消耗品有关的条文
4管理要求
4.2质量管理体系
4.2.4质量手册应对质量管理体系及其所用文件的架构进行描述。医学实验室质量手册的目录可包括以下内容:
a) 引言;
……
i) 仪器,试剂和/或相关消耗品的管理;
…….
4.2.5实验室管理层应建立并施行一个计划,用于定期监控和证实仪器、试剂及分析系统经过了适当校准并处于正常功能状态;还应有一套记录在案的预防性维护及校准(见5.3.2)文件,其内容至少应遵循制造商的建议。
4.6外部服务和供应
4.6.1实验室管理层应建立并文件化其政策和程序,以选择和使用所购买的可能影响其服务质量的外部服务、设备以及消耗品等。所购买的各项物品应符合实验室的质量要求。国家、地区或地方法规可能要求保存采购物品的记录。应该有对消耗品进行检查、接受/拒收和贮存的程序和准则。
4.6.2采购的设备及消耗品可能会影响服务质量时,在确定这些物品达到标准规格或有关程序中规定的要求之前,不得使用。可通过检验质控样品并验证结果的可接受性而做出决定。还可利用供应商对质量管理体系的符合性声明进行验证。
4.6.3应建立一套供货清单控制系统。应按照质量管理体系的规定,对外部服务、供应物品以及所购买的产品建立适当的质量记录并保持一定的时间。该系统中应该包括全部相关试剂、质控材料以及校准品的批号、实验室接收日期以及这些材料投入使用的日期。所有这些质量记录应在实验室管理评审中提供。
4.6.4实验室应对影响检验质量的重要试剂、供应品以及服务的供应商进行评价,并且保存这些评价的记录和经批准的清单。
4.9不符合项的识别和控制
4.9.1当发现检验过程的任何方面有不符合所制定的程序或质量管理体系的要求,或不符合申请检验的临床医生的要求时,实验室管理层应有相应的政策和程序并实施,以确保:
注:不符合的检验或操作活动可以出现在不同的方面并有不同的识别方法,包括医生的投诉、质量控制提示、设备校准、消耗品检查、工作人员的意见,报告和证书的检查,实验室管理层的评审,以及内部和外部审核等。
5 技术要求
5.2设施和环境条件
5.2.9应提供相应的存储空间和条件,以保证样品、切片、组织块、保存的微生物、文件、手册、设备、试剂、实验室用品、记录以及检验结果等的完整性。
5.3实验室设备
注:在本标准中,仪器设备、参考物质、消耗品、试剂和分析系统等也包括在实验室设备中(可行时)。
5.3.2应确定设备(在安装时及常规使用中)能够达到所要求的性能标准,并且符合相关检验所要求的条件。
实验室管理层应建立一套程序,用于定期检测并证实设备、试剂、分析系统等经过了适当校准并处于正常功能状态。同时还应有记录并归档的预防性维护程序(见4.2.5),该程序至少应遵循制造商的建议。
如果有制造商的使用说明、操作手册或其它相关文件,可酌情使用这些资料建立符合相关标准的要求,明确校准周期,以满足本款的部分或全部要求。
5.3.14包括硬件、软件、质控物质、消耗品、试剂和分析系统在内的设备均应得到相应的保护,以避免造成导致检验结果无效的调整或改动。
5.5.3所有的程序都应形成文件,使相关工作人员可以从工作站上查阅。已形成文件的程序及必要的指导书应使用实验室工作人员都理解的语言。
卡片文件或类似总结有关键信息的系统可供工作人员在工作台上快速查阅,同时应备有完整的操作手册供检索。卡片文件或类似的系统应与完整手册的内容相对应。任何节略性的程序都应该作为文件控制系统的一部分。
如果制造商提供的使用说明书(如包装插页)符合5.5.1和5.5.2的要求,其描述可作为实验室操作的程序,所使用的语言能被实验室的工作人员所理解,则检验程序应部分或全部地以此说明书为基础来制定。任何偏离均应经过评审并形成文件。进行检验所需的附加信息也应形成文件。每个新版检验试剂盒在试剂或程序方面发生重大变化时,均应进行性能和适用性检查。与其它程序一样,任何程序上的变化都应注明日期并经授权。
除了文件控制标识,文件中还应包括以下内容(当可行时):
a) 检验目的;
b) 用于检验程序的原理;
c) 性能参数(如线性、精密度、以测量不确定度表示的准确性、检出限、测量区间、测量真实性、灵敏度和特异性);
d) 原始样品系统(如血浆、血清和尿液);
e) 容器和添加剂类型;
f) 所需设备和试剂;
g) 校准程序(计量学溯源性);
h) 程序步骤;
i) 质量控制程序;
j) 干扰(如乳糜血、溶血、胆红素血)和交叉反应;
k) 结果计算程序的原理,包括测量不确定度;
l) 生物参考区间;
m) 患者检验结果的可报告区间;
n) 警告/危急值(当适用时);
o) 实验室解释;
p) 安全性预警措施;
q) 变异的潜在来源。
只要具备上述信息,也可以使用电子手册的形式。对于文件控制的要求同样适用于电子手册。
实验室负责人应负责保证检验程序内容的完整和现行有效,并进行全面的评审。
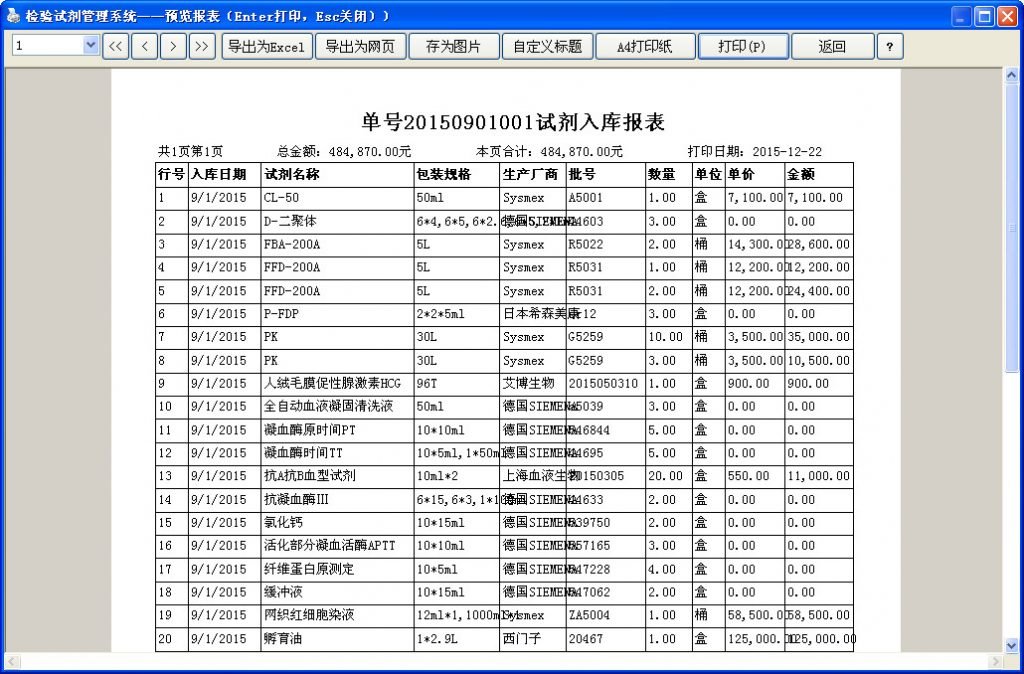
相关链接:下载试用检验试剂管理系统